
Contents
- What is Pfeiffer syndrome?
- Pfeiffer syndrome types
- What are the causes of Pfeiffer syndrome?
- Pfeiffer syndrome diagnosis
- Pfeiffer syndrome treatment
- Minimally invasive skull repair for Pfeiffer syndrome
- Fronto-orbital advancement for Pfeiffer syndrome
- Le Fort advancements for Pfeiffer syndrome
- Distraction osteogenesis for Pfeiffer syndrome
- Airway treatments for Pfeiffer syndrome
- Dental Treatment for Pfeiffer syndrome
- Surgery to correct finger and toe problems in patients with Pfeiffer syndrome
- Hearing issues treatment for Pfeiffer syndrome
- Pfeiffer syndrome survival rate
What is Pfeiffer syndrome?
Pfeiffer syndrome is very rare (1:100 000 births) genetic, dominantly inherited disorder characterized with craniosynostosis which is a medical term for premature fusion of certain skull bones.
Too early fusion of skull bones may prevent skull normal growth affecting the shape of a face and head. Abnormalities of skull bones may lead to high forehead, bulging and wide-set eyes, underdeveloped upper jaw and a beaked nose. More than 50% of all patients with Pfeiffer syndrome have hearing loss. Dental problems are also common.
This syndrome may also affect bones in the hands in feet causing abnormalities such as wide and deviated thumbs and toes. The thumbs and toes are wide and bend away from the other digits. Brachydactyly or unusually short fingers and toes and syndactyly or webbing or fusion between the digits are also very common.
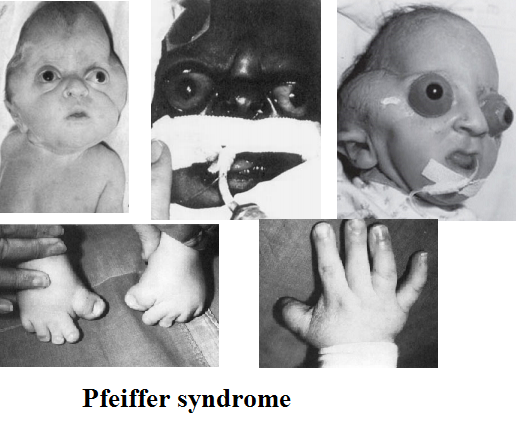
What are different names of Pfeiffer syndrome?
The disease is also commonly known as Craniosynostosis. Other names are:
- Acrocephalosyndactyly, type V
- ACSV
- Craniofacial-skeletal-dermatologic syndrome
- Noack syndrome
Pfeiffer syndrome types
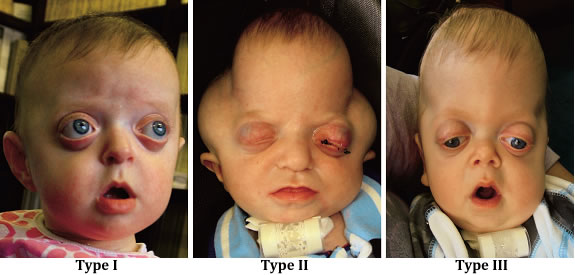
According to M. Michael Cohen classification Pfeiffer syndrome can be divided into 3 types characterized by broad thumbs, broad great toes, brachydactyly, and possibly syndactyly. These types can possibly overlapping.
- Pfeiffer syndrome Type 1 or classic Pfeiffer syndrome is characterized by craniosynostosis and “midface deficiency”. Patients with this type inherited the disease in an autosomal dominant pattern. Most patients with Pfeiffer syndrome type 1 have normal intelligence and a normal life span.
- Pfeiffer syndrome Type 2 is characterized by a cloverleaf-shaped skull (Kleeblattschädel). This is caused due to extensive bones fusion and also because of severe proptosis. This type does not appear to be inherited and occurs sporadically and has severe neurological issues unfortunately a poor prognosis, generally with early death.
- Pfeiffer syndrome Type 3 is characterized by craniosynostosis and severe proptosis. This type also occurs sporadically. It does not appear to be inherited and also has a poor prognosis.
The early skull bones fusion can limit brain growth and development, leading to various neurological problems. Moreover, patients with type 2 or 3 Pfeiffer syndrome can have ankylosis – fusion of the bones in the elbow or other joints, which may limit mobility, and abnormalities of the face and respiratory tract provoking life-threatening breathing problems.
The main difference between type 2 and type 3 is the presence of a cloverleaf-shaped head, which is caused by more extensive skull bones fusion.
What are the causes of Pfeiffer syndrome?
Pfeiffer syndrome is a genetic autosomal dominant disease. Dominant genetic disorders occur when only a single copy of an abnormal (e.g. mutated) gene is needed to precipitate a particular disease. In case of for Pfeiffer syndrome type I, the abnormal gene is inherited from either parent, whereas Pfeiffer syndrome type II and type III are the result of a new mutation in the affected individual.
Studies have been shown that the older the father is the higher are chances for new mutations causing Pfeiffer syndrome. The risk that abnormal gene will pass from an affected parent to a child is 50% for each pregnancy. The risk is the same for males and females.
Pfeiffer syndrome is usually caused by mutations in the fibroblast growth factor receptor – FGFR2 gene. Mutations in the FGFR1 gene cause a smaller number of cases of type 1 Pfeiffer syndrome. Gene mutations have not been related with type 2 or 3. The FGFR1 and FGFR2 genes are needed to provide appropriate instructions for the production of receptors known as fibroblast growth factor receptors 1 and 2.
During embryonic development these proteins signals are needed for the transformation of immature cells into bone cells. A mutation in either the FGFR1 or FGFR2 gene alters the function of the described protein, provoking prolonged signaling, promoting the premature fusion of skull, hand and feet bones.
It should be known that Pfeiffer syndrome is not the only one known disorder linked with FGFR mutations. Other disorders are also linked with FGFR gene mutations including: Apert syndrome, Beare-Stevenson syndrome, Crouzon syndrome, Jackson-Weiss syndrome, and FGFR2-related isolated coronal synostosis, Crouzon syndrome with acanthosis nigricans and Muenke syndrome.
What are signs and symptoms of Pfeiffer syndrome?
Signs and symptoms of Pfeiffer syndrome may vary depending on its type as described below:
Signs and symptoms of Pfeiffer syndrome type 1
Infants with Pfeiffer syndrome type I have or may have following signs and symptoms:
- Craniosynostosis which causes the head to appear short and tall, medically also known as turribrachycephaly
- A high, full forehead
- Midface hypoplasia with underdeveloped midfacial regions
- Ocular hypertelorism with widely spaced eyes
- Hypoplastic maxilla with underdeveloped upper jaw with a prominent lower jaw
- Dental abnormalities
- Intelligence and life span are usually normal.
Signs and symptoms of Pfeiffer syndrome type 2
Individuals with Pfeiffer syndrome type II usually have more severe signs and symptoms including:
- Severe form of craniosynostosis known as Cloverleaf skull with more severe hand and foot abnormalities and additional malformations of the limbs. Kleeblattschadel type craniosynostosis causes the skull to have a “tri-lobed” appearance.
- Craniosynostosis is often linked with hydrocephalus, a condition with altered cerebrospinal fluid – CSF that may lead to abnormal widening of the spaces within the brain causing accumulation of CSF in the skull and increased pressure on the brain.
- Abnormally high and broad forehead
- Ocular proptosis – severe protrusion of the eyes
- Midface hypoplasia – flat middle portion of the face
- “Beak-shaped” nose
- Downwardly displaced ears
- Ankylosis – lack of mobility of the elbow joints
- Visceral organs anomalies
- Impaired mental development and neurological problems
- Respiratory issues
- Hypoxia
Signs and symptoms of Pfeiffer syndrome type 3
Individuals with Pfeiffer syndrome type III usually have similar signs and symptoms with to those with Pfeiffer syndrome type II with the exclusion of the cloverleaf skull deformity. Additional signs and symptoms may include:
- Anterior cranial base – shortened base of the skull
- Natal teeth – the abnormal presence of certain teeth at birth
- More severe protrusion of the eyes
- Viscelar anomalies
- Impaired mental development and neurological problems
Pfeiffer syndrome diagnosis
The diagnosis is confirmed according to clinical findings. Molecular genetic testing for FGFR1 and FGFR2 is available if the diagnosis is uncertain.
Differential diagnosis should be sometimes made between Pfeiffer, Apert, Muenke, and Beare-Stevenson syndromes. Apert syndrome and Pfeiffer syndrome are notable for some similarities but the two disorders are etiologically and genetically very distinct. Crouzon and Pfeiffer syndromes are phenotypically similar but in Crouzon syndrome, hand and foot anomalies are lacking.
Phenotypic overlap may between Pfeiffer and Muenke syndrome, which is caused by a specific FGFR3 mutation. Sometimes Pfeiffer syndrome has been confused with Jackson-Weiss and Saethre-Chotzen syndromes, since broad toes may occur in both.
Can Pfeiffer syndrome be diagnosed prenatal?
The syndrome is usually noticed in the newborn period or later, and in very rare cases prenatally. Prenatal diagnosis has only been confirmed 6 times, mostly because of the presence of a cloverleaf skull deformity. A careful 3D ultrasound examination may lead to an early prenatal diagnosis also in cases without cloverleaf skull.
The large clinical manifestation variability of this disorder even within the same family, but also with other causes of craniosynostosis might prenatal sonography diagnosis alone difficult. Thus consequent molecular analysis should be performed to confirm the diagnosis by identifying a FGFR mutation.
Pfeiffer syndrome treatment
Pfeiffer syndrome is a complex disorder and usually involves several parts of newborn body, most commonly: face, head and limbs. Because this condition also involves the skull and face, it may also affect child’s hearing, speech, feeding and dental functions. Your child may need several treatments and surgeries to repair these anomalies.
These procedures may be very successful. Some of the following treatments options are described in further text:
Minimally invasive skull repair for Pfeiffer syndrome
This surgery is performed by releasing parts of tissue that connect the skull and bones, called sutures, through several small incisions, as a replacement for of traditional open surgery.
If it is performed during the first few months of a newborn’s life, this procedure may help to correct the skull shape and provides space for the brain to grow. After the procedure is done, child will be fitted with a special, temporary helmet in order to correct the skull shape.
Fronto-orbital advancement for Pfeiffer syndrome
If minimally invasive skull repair is not an adequate option for the newborn, fronto-orbital surgery may be performed to correct the skulls when your child is between 9 and 11 months of age. This procedure is characterized by removing or repairing the fused sutures and reshaping the skull. In this procedure zigzag incision technique is used which may help to camouflage the scars and makes the incision less visible.
Le Fort advancements for Pfeiffer syndrome
Pfieffer syndrome is also present with upper jaw and teeth abnormalities. These types of surgeries of the upper jaw may correct abnormalities of the maxilla (upper jaw), including the way the teeth align.
Procedure includes sectioning or cutting and repositioning of the upper jaw in order to correct its irregular position. 3D imaging technology is also used in Le Fort procedures, which may enable surgeons to create a precise surgical plan.
Distraction osteogenesis for Pfeiffer syndrome
This procedure moves two bone segments gradually apart so that new bone may be able to fill in the gap. During this procedure, a surgeon makes a break or osteotomy in the anomalous bone and attaches the device called a distractor to both sides of the osteotomy.
This distractor is then slowly adjusted over a period of days to stretch the gap and enable new tissue to fill it in. This procedure is often done after the Le Fort advancement procedure.
Airway treatments for Pfeiffer syndrome
Newborns with Pfeiffer syndrome may have blocked airways, especially in those with Pfeiffer syndrome type 2 and 3. Airway obstructions can be treated in many ways. Some advanced treatments may include:
- Tonsillectomy and or adenoidectomy
- Wearing a continuous positive airway pressure (CPAP) mask
- Midfacial advancement also known as “Le Fort III”
- In severe cases, placement of a tracheostomy may be needed until the newborn gets old enough to undergo other operations.
Dental Treatment for Pfeiffer syndrome
Pfeiffer syndrome may also affect the position of the teeth and the fitting together of the upper and lower teeth when the jaw is closed. Many newborns with Pfeiffer syndrome may have an under bite, but also misaligned teeth. Delayed tooth eruption may also be present. Pediatric dentist specialist should be consulted when the teeth start to come in but no later than 2 to 3 years of age.
Surgery to correct finger and toe problems in patients with Pfeiffer syndrome
In patients with Pfeiffer syndrome, the thumbs and the big toes are usually wider than normal. This problem may not require any treatment, but it is one of the major markers to make the precise diagnosis of Pfeiffer syndrome. Although it is rare, syndactyly – fusion of the fingers or toes may also be caused. It is recommended that syndactylies should be be separated, and this is best done at experienced centers.
This separation can sometimes be synchronized with skull surgery, or other surgeries, in order to reduce the total number of operations.
Hearing issues treatment for Pfeiffer syndrome
In the cases of Pfeiffer syndromes, ears are usually normally shaped, but they are positioned slightly lower than normal. As a result of mid-facial hypoplasia the inner ear may not drain well, and children commonly develop otitis media, or ear infections.
Most often, children need placement of ear small tubes to prevent these infections and chronic scarring of the eardrum. In some cases, the inner ear bones may be partially fused which may lead to the conductive hearing loss.
It has been shown that almost 60% of all children with Pfeiffer syndrome have incompletely formed ear canals. In the case of type 3 Pfeiffer syndrome, almost all children have such issue. BAHA’s – bony conductive hearing aids that are implanted into the skull may be effective treatment for such children.
However it should be delayed until children are in their teens, because the presence of BAHA’s can complicate some skull surgeries that are needed before adolescence. Many adults with Pfeiffer syndrome have some degree of hearing loss, thus careful monitoring to the ears is important in infancy and childhood.
Pfeiffer syndrome survival rate
Timely treatment and its success is the main prognostic factor of Pfeiffer. Patients with Pfeiffer syndrome type 1 usually have normal life span compared to those with type 2 and 3. Pfeiffer syndrome is rarely encountered, even at well-known craniofacial centers. Both Pfeiffer syndrome types II and III are related with a greater risk of death early in life, and some centers have reported survival rates from 15% to 75% in childhood.
Favorable outcomes can be attained but the prognosis still remains poor in severe cases because of the risk of respiratory and neurological complications.
Many health professionals believe that a preemptory tarsorrhaphy can prevent to early visual loss and that further elevation of survival rates may be possible with aggressive airway strategies including early tracheostomies and more frequent screening such as magnetic resonance imaging for Chiari malformations.
“Angular cheilitis: Causes, symptoms, treatment and pictures“